WALNUT CREEK, CA—Today’s powerful sequencing machines can rapidly read the genomes of entire communities of microbes, but the challenge is to extract meaningful information from the jumbled reams of data. In a paper appearing in Nature Biotechnology August 17, a collaboration headed by researchers at the University of Washington and the U.S. Department of Energy Joint Genome Institute (DOE JGI) describes a novel approach for extracting single genomes and discerning specific microbial capabilities from mixed community (“metagenomic”) sequence data.
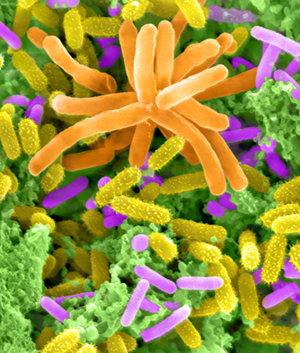
Methylamine-enriched community of Lake Washington sediment featuring Methylotenera cells. Photo © Dennis Kunkel Microscopy, Inc. (Color by Ekaterina Latypova)
For the first time, using an enrichment technique applied to microbial community samples, the research team explored the sediments in Lake Washington, bordering Seattle, WA and characterized biochemical pathways associated with nitrogen cycling and methane utilization, important for understanding methane generation and consumption by microbes. Methane is both a greenhouse gas and a potential energy source.
“Even if you have lots of sequence, for complex communities it still doesn’t tell you which organism is responsible for which function,” said the paper’s senior author Ludmila Chistoserdova, a microbiologist at the University of Washington. “This publication presents an approach, via simplification and targeted metagenomic sequencing, of how you can go after the function in the environment.”
Chistoserdova and colleagues study microbes that oxidize single-carbon compounds such as methane, methanol and methylated amines, which are compounds contributing to the greenhouse effect and are part of the global carbon cycle.
“To utilize these single-carbon compounds, organisms employ very specialized metabolism,” said Chistoserdova. “We suspect that in the environment, there are novel versions of this metabolism, and possibly completely novel pathways.”
Most of the microbes that oxidize single-carbon compounds are unculturable and therefore unknown, as are the vast majority of microbes on Earth. To find species of interest, the researchers sequenced microbial communities from Lake Washington sediment samples, Chistoserdova said, because lake sediment is known to be a site of high methane consumption. However, these sediment samples contained over 5,000 species of microbes performing a complex, interconnected array of biochemical tasks.
To enrich the samples for the microbes of interest, the researchers adapted a technique called stable isotope probing. This is the first time the technique has been used on a microbial community, Chistoserdova said. The researchers used five different single-carbon compounds labeled with a heavy isotope of carbon, and fed each compound to a separate sediment sample. The microbes that could consume the compound incorporated the labeled carbon into their DNA, Chistoserdova said, while organisms that couldn’t use the compound did not incorporate the label. The labeled DNA was then separated out and sequenced. In this way, microbial “subsamples” were produced that were highly enriched for organisms that could metabolize methane, methanol, methylated amines, formaldehyde and formate.
The functionally enriched samples contained far fewer microbes than the total sample, Chistoserdova said. The sample that was fed methylated amines was simple enough that the group was able to extract the entire genome of a novel microbe, Methylotenera mobilis, that normally comprises less than half a percent of the community, but appears to be a first responder to methylated amines in the environment. The researchers were able to construct much of M. mobilis’ biochemistry, and predict that it is also involved in nitrogen cycling, demonstrating the utility of metagenomic analysis.
The DOE JGI performed the sequencing and assembly of these complex metagenomic data sets. The complexity of the community’s sequence samples created new challenges for genome assembly. “It is very important for metagenomic assemblies to rely on high-quality reads,” said Alla Lapidus, microbial geneticist at the DOE JGI and co-author on the paper. If some of the sequence is of low quality, she said, it can lead to errors in assembly and gene annotation.
Because of the need for higher quality control, Lapidus said, the DOE JGI developed a new quality control approach that involves a computer tool called LUCY to trim out low-quality sequence in combination with the Paracel Genome Assembler, which appeared to be more appropriate for metagenomic assemblies. This approach was pioneered on the Lake Washington project, Lapidus said, and due to its superior results it is now the standard metagenomic assembly method at the DOE JGI.
“The DOE JGI’s unique Integrated Microbial Genomics with Microbiome Samples (IMG/M) [http://img.jgi.doe.gov/m] data management system was used for detailed annotation, and was instrumental for efficient comparative analysis and metabolic reconstruction of the samples,” Lapidus said.
Michael Galperin, a microbial geneticist at the National Center for Biotechnology Information at the National Institutes of Health, who was not involved in the study, said in an email that the paper describes “an interesting novel approach” and the results “constitute a significant advance in the emerging discipline of metagenomics.”
“I think other people can use the same approach in different environments, as long as they have an enrichment technique,” Chistoserdova said. “For us this work is just the beginning, because now we will be using this metagenomic sequence as a scaffold for downstream experiments in our lake.”
Other DOE JGI authors include Natalia Ivanova, Alex Copeland, Asaf Salamov, Igor Grigoriev, Susannah Tringe, David Bruce (Los Alamos National Laboratory) and Paul Richardson; and Ernest Szeto and Victor Markowitz of the Data Management and Technology Center, Lawrence Berkeley National Laboratory.
The U.S. Department of Energy Joint Genome Institute, supported by the DOE Office of Science, unites the expertise of five national laboratories — Lawrence Berkeley, Lawrence Livermore, Los Alamos, Oak Ridge, and Pacific Northwest — along with the Stanford Human Genome Center to advance genomics in support of the DOE missions related to clean energy generation and environmental characterization and cleanup. DOE JGI’s Walnut Creek, CA, Production Genomics Facility provides integrated high-throughput sequencing and computational analysis that enable systems-based scientific approaches to these challenges.