The main exercise dealt with only one dimension of assembly–the sequence. In addition to the sequence, the quality of the sequence also must be considered.

Click the image to see a larger version.
The trace image is from a capillary sequencer. The peaks represent fluorescently labeled fragments that passed the reader at a point in time. Taller peaks represent large quantities of fragments that migrated past the reader at that instance. Shorter peaks represent fewer fragments. When peaks are taller and distinct, they are assigned a high quality score (e.g., the A peak at position 449). In addition, homopolymer regions (runs of the same base in a row) can be problematic. Look at the run of 3 Gs from 453 to 455. This section is not problematic, as each of the 3 Gs has its own distinct peak. Now look at the run of three Gs from 470 to 472. This is one large, spread-out peak. The large peak obviously contains more than one G, but how many does it contain? So the quality for this section would be lower.
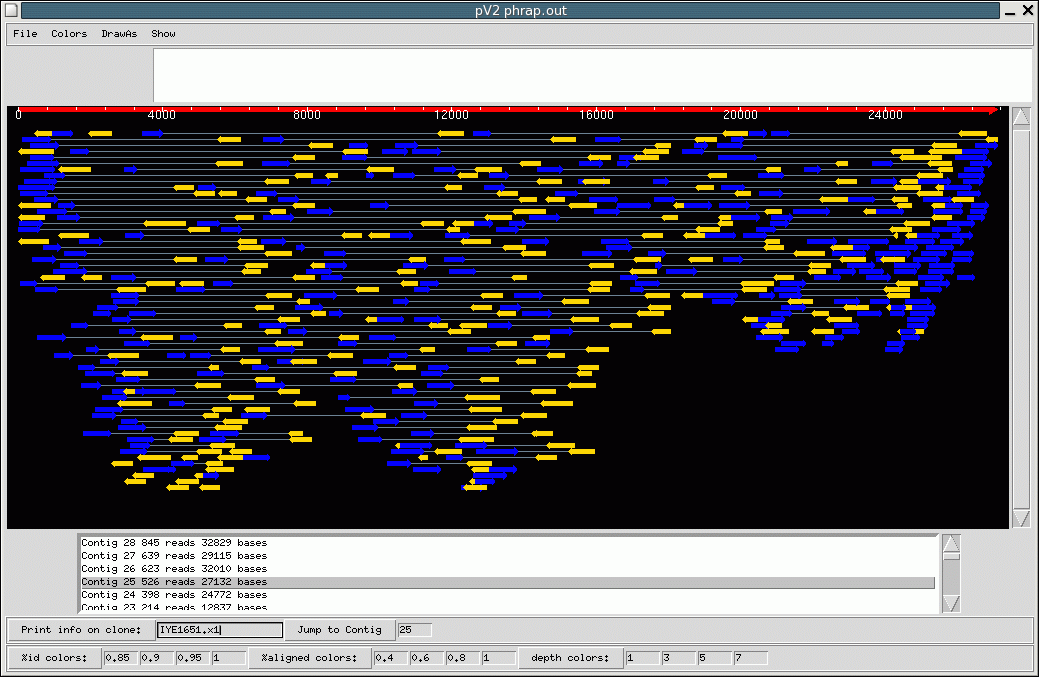
Click the image to see a larger version.
The phrap image shows an assembly of many different paired reads. The blue arrows represent sequence results from the sense strand, and the yellow arrows are the antisense strand. The thin grey line between them represents the unsequenced bases (Ns) between the two sequencing reactions. This image illustrates the number of reads required to assemble a 28,000-bp section of DNA.
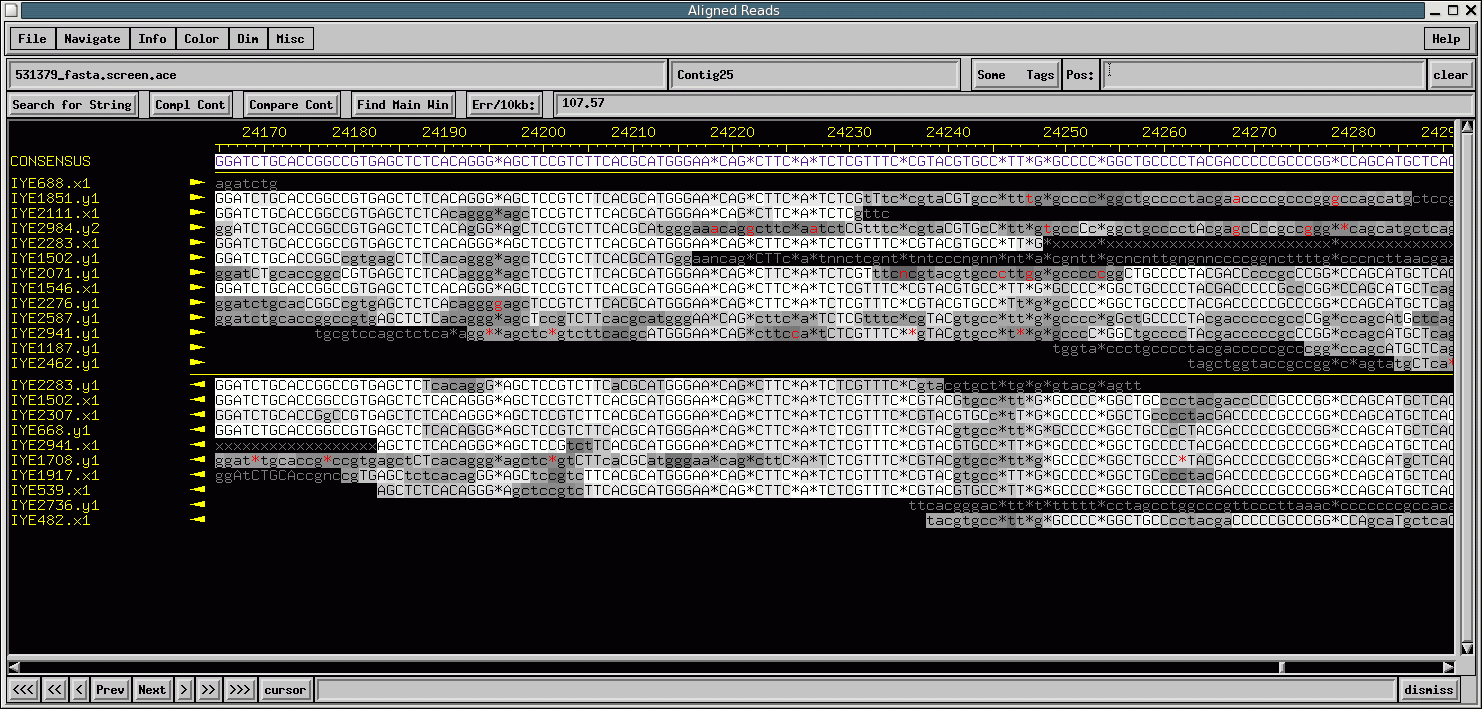
Click the image to see a larger version.
The consed image illustrates depth of coverage and quality. The top line is the consensus sequence. This sequence is created from the consensus of all the underlying sequences below. Each row below represents results of a single sequencing reaction. As you can see, we have as much as 18x depth of coverage in areas of this assembly. The lighter the background for the base, the higher the quality of that base. If a letter’s background is dark and lower case, the quality of the base is poor. By using depth of coverage and quality, we can make educated decisions on the sequence when conflicts exist amongst reads. For example, from position 24193 to 24195 there is a run of three Gs. 16 of the 18 reads say there are three Gs. One low-quality read says there are four, and another low-quality read says there are two. Many of the 16 reads have good quality. Based on this information, one should conclude that the consensus sequence has three Gs. The *s in the sequence are space holders in a sequence so that the rest of the reads line up after there has been an insertion or deletion. Note the consensus sequence with the * after 24195 does not actually have a marker counting the base position. Red bases are the actual bases for the insertion or deletion.