Using a novel computational approach, researchers confirm microbial diversity is wilder than ever.
Imagine researchers exploring a dark room with a flashlight, only able to clearly identify what falls within that single beam. When it comes to microbial communities, scientists have historically been unable to see beyond the beam — worse, they didn’t even know how big the room is.
A new study published online Oct. 11, 2023 in Nature highlights the vast array of functional diversity of microbes through a novel approach to better understand microbial communities by looking at protein function within them. The work was led by a team of scientists at the U.S. Department of Energy (DOE) Joint Genome Institute (JGI), a DOE Office of Science User Facility located at Lawrence Berkeley National Laboratory (Berkeley Lab), and collaborators across multiple other research centers around the world.
“We’ve more than doubled the number of protein families known up until now, and identified many novel structure predictions,” said lead author on the paper Georgios Pavlopoulos, now a research director at the Biomedical Sciences Research Center Alexander Fleming. “This was a massive analysis of 1.3 billion proteins with massively parallel computations.”
Guided by JGI scientists, the team embarked on a mission to unveil the mysteries concealed within the “dark” functional realm. Their focus sharpened on deciphering the intricate world of protein functional diversity: the novel protein families and novel functions in as-yet unveiled microbes. Harnessing the collective power of more than 26,000 microbiome datasets, all accessible through the publicly available Integrated Microbial Genomes & Microbiomes (IMG/M) database, they successfully crafted the Novel Metagenome Protein Families (NMPF) Catalog.
“We can now analyze new datasets by comparing against these protein families, or further analyze the protein families in order to predict new functions,” said Nikos Kyrpides, senior author of the study and head of the JGI’s Microbiome Data Science group.
Shining a Light on Functional ‘Dark Matter’
Microbial communities living everywhere from soils and stomachs to the deep sea are capable of doing a lot of unique things when it comes to energy cycles — turning biomass into things like ethanol or hydrogen, or solar energy into hydrogen.
Microbial communities are also incredibly difficult to study. Many of the microbes within them cannot be cultivated in lab settings. Since each microbial community has its own unique makeup of microbe players and the functions they perform, artificially replicating a whole community is impossible.
Metagenomic sequencing allows researchers to study the entire genetic makeup of these communities via whole genome sequencing of the samples, without being able to distinguish which gene belongs to each individual microbial species within a community. Therefore, the process hinges on referencing to existing genome sequences.
Some of these proteins are what the scientists call “known knowns” — that is, they are similar to genes with known function. Others are called ‘known unknowns’ — that is, they are similar to previously known genes from isolate organisms, but we still aren’t sure of their function.
However, if a gene in the community doesn’t match any of the previously known genes from isolates, there isn’t much scientists can tell about its function or its origin. As a result, these genes were typically discarded from any analysis as useless information. These represent the ‘unknown unknowns’ because they aren’t similar to anything we’ve already defined.
“A huge percentage — around 30–50% of the protein families that we knew so far — still does not have any known function, but we knew the families,” Kyrpides said. Yet, “almost 20 years of metagenomic data and metagenomic analysis, and still there has been no real analysis of protein families from metagenomes per se.”
Recently, other research teams have leveraged the power of artificial intelligence to decode the language of protein sequences and obtain hints of their possible functions. Yet these efforts were limited to the realm of already-known protein sequences.
“In this endeavor, we have not only ventured into the uncharted territory of understanding the vast landscape of functional diversity, but we have also pushed the boundaries by applying AI methodologies to unravel their roles,” Pavlopoulos said. “Consequently, we have amassed an extensive repository of groundbreaking insights, significantly expanding the horizons of potential functions across various categories of proteins, including those with pivotal applications in biotechnology, such as DNA editing enzymes.”
Leveraging Protein Families in a New Way
The discovery of new protein families had started to plateau in recent years, perhaps suggesting that scientists had “captured” much of the diversity out there, even if it hadn’t yet defined what it did, exactly. But what kind of diversity might those ‘unknown unknowns’ hold?
The team started with 8 billion metagenome genes from IMG (the study also references data from the JGI’s Genomes from Earth’s Microbiome, or GEM catalog). Then they removed any genes with even a remote similarity to previously known genes, leaving them with around 1.2 billion novel genes.
They took what they were left with and clustered them into families. From there they focused on families with at least 100 members.
“If you have 100 sequences, the quality of the cluster is significantly higher because it is very hard to have 100 sequences from different locations or habitats that align very well, randomly,” Kyrpides explained. “Replicating that 100 times would have been almost impossible.”
When the team was finished with this phase, they found that the protein family diversity within this metagenomic space (the ‘unknown unknowns’) was vastly greater than that of the reference genomes — by at least double.
“As we keep on adding more samples, we’re getting more protein families,” Kyrpides said. “In a few years, as we keep on sequencing more metagenomes, some of the clusters that have currently 50 members or more will grow to 100 members or more as well. So, we’re saying diversity has doubled, but in reality it could be three or four or five or tenfold more out there.”
Digging Further into an Array of Diversity
While the team didn’t drill down function, they were able to further characterize these families. They divided the protein families up by environment and found only 7% of protein families were shared across all eight environmental categories. Instead, families preferred a specific environment — whether that be soil, animal hosts, marine ecosystems, etc.
“So, they must be doing something interesting or important for that habitat,” Pavlopoulos explained. “That is definitely material that the scientific community now can use further. Let’s say somebody is working on soil environments or the human body — they may take some of those families and try to functionally characterize them because they are very specific to that habitat.”
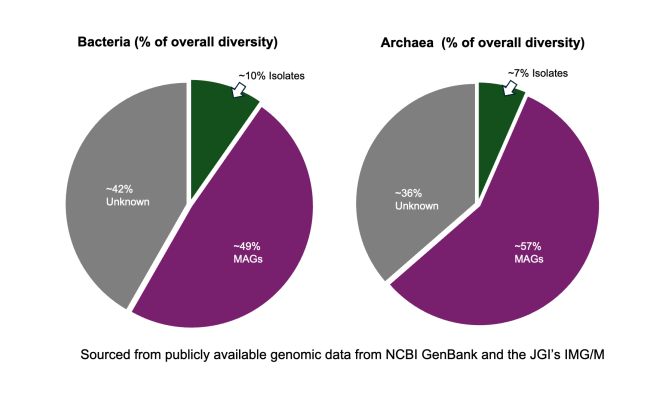
Taxonomic analysis found that the majority of these protein families belonged to bacteria and viruses, though 6 million of the sequences evaded classification. Researchers also tried to hone in on the function of the genes via 3D modeling, and comparing structures of the unknown to those of the known — similar structure equates to high likelihood of similar function. The team also identified protein families with completely novel structures. The computational power to perform this level of analysis hinged on access to the National Energy Research Scientific Computing Center, another user facility at Berkeley Lab.
“It’s also a credit to Aydin Buluç’s team with Berkeley Lab’s Applied Mathematics and Computational Research Division,” Pavlopoulos said. “They developed parallel algorithms to perform ‘all-vs-all’ comparisons and graph clustering able to run in such highly parallel infrastructures.”
This is the first time protein structures have been used to help characterize the vast array of microbial dark matter. The study took roughly two years to complete, with only about 20,000 metagenomes sequenced at the time. Now, that number is closer to 60,000.
“There is still 70–80% of known microbial diversity out there that is not yet captured genomically,” Kyrpides said. “So, that diversity is definitely holding a lot of new secrets in terms of functional diversity as well.”
Researchers from Harvard University, Indiana University. University of Crete (Greece). Georgia Institute of Technology, Michigan State University, Lawrence Livermore National Laboratory, University of Washington, Centre for Research & Technology Hellas (Greece), Aristotle University of Thessalonica (Greece), and the University of California, Berkeley were also involved in the work. Other authors on the paper are Fotis Baltoumas, Sirui Liu, Oguz Selvitopi, Antonio Camargo Stephen Nayfach, Ariful Azad, Simon Roux, Lee Call, Natalia N. Ivanova, I Min Che, David Paez-Espino, Evangelos Karatzas, Novel Metagenome Protein Families Consortium, Ioannis Iliopoulos, Konstantinos Konstantinidis, James M. Tiedje, Jennifer Pett-Ridge, David Baker, Axel Visel, Christos A. Ouzounis, and Sergey Ovchinnikov.

More Details
Metagenome
Microbial
Viral
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Director's Science, R&D, and Work For Others
Bioenergy Research Centers
Bioenergy Research Centers
Bioenergy Research Centers
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Community Science Program
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
Director's Science, R&D, and Work For Others
ETOP
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Director's Science, R&D, and Work For Others
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Director's Science, R&D, and Work For Others
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Director's Science, R&D, and Work For Others
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Director's Science, R&D, and Work For Others
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
BER Support Science
Community Science Program
Community Science Program
Community Science Program
BER Support Science
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Other
Other
Community Science Program
Community Science Program
Bioenergy Research Centers
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Director's Science, R&D, and Work For Others
Community Science Program
Bioenergy Research Centers
Bioenergy Research Centers
Bioenergy Research Centers
Community Science Program
Community Science Program
Bioenergy Research Centers
Community Science Program
Director's Science, R&D, and Work For Others
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Community Science Program
Bioenergy Research Centers
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
EMSL FICUS
Pavlopoulos, G. A. et al. (2023) Unraveling the functional dark matter through global metagenomics. Nature. 10.1038/s41586-023-06583-7


Metagenome Program
The Metagenome Program uses advanced sequencing technologies to decode the genomes of microbes in their natural environments.